

Back to top
Whole Genome Sequencing

- Detect single nucleotide to large structural variations in your model organism
- Detect off target mutations of your CRISPR/Cas9 gene editing experiment
- validate your bacterial production strain
- characterize microbial isolates by MLST
Overview
Considerations before starting a whole genome sequencing project:
- Scientific objective
- Availbility and quality of a reference genome
- SNV detection and/or SV detection
- Optimal coverage/sequencing depth & read length
- Are substantial DNA contaminations suspected
Let us guide you – from design to analysis
Example projects using whole genome sequencing:
- Detection of mutations aquired by cancer cells – from SNPs to large structural variations
- Insertion site detection
- Mutation verification, ruling out off-target mutations
- Genetic modifications in breeding studies
Applications related to whole genome sequencing:
- RNA Sequencing
- Whole exome resequencing
Workflow
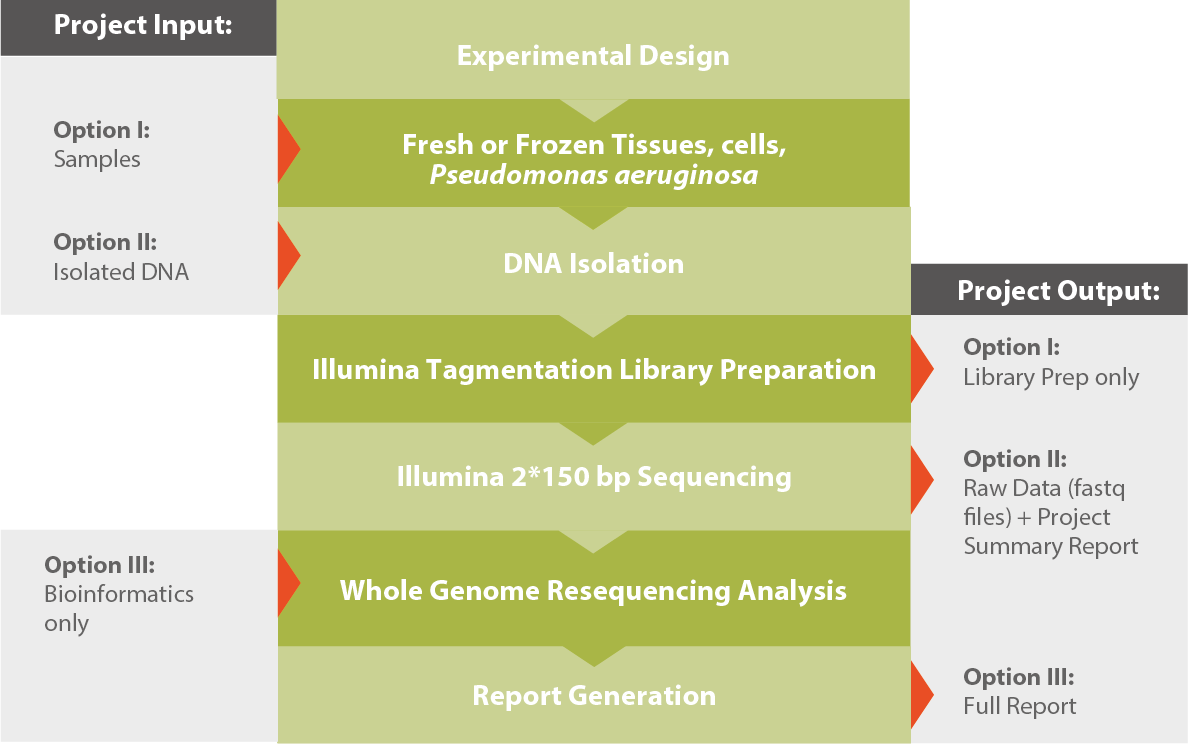
Results
Without Bioinformatics
Raw data:
If no analysis module is ordered, Microsynth provides for Whole Genome Sequencing the key outcomes as listed below:
- Assessment of Sequencing Quantity and Quality(in .xlsx format)
- The evaluation of the quantity and quality of the sequencing data.
- Raw data ( per sample, in .fastq format)
- The raw data allows you to run your own analysis or track back to each nucleotide sequenced.
- A Project summary report(.pdf format)
- The report summarises the key Parameters of the Project.
With Bioinformatics
Standard Bioinformatic Analysis:
For our resequencing application, in addition to the raw data, Microsynth's analysis module provides a variety of insights to meet your scientific objectives:
- Sequencing Quantity and Quality Evaluation (in .xlsx and .html format)
A comprehensive assessment of sequencing quantity and quality, providing crucial insights. - Alignment/Map Files and Indexes (in .bam and .bai format)
Access alignment and map files along with their corresponding indexes for your convenience. - Genome Coverage and Read Depth Analysis(in .tsv and .bed format; see Figure 1)
Gain insights into genome coverage and read depth through results provided in .tsv and .bed formats. - Structual variations & Copy number analysis (compare Table 1 & Figure 2)
- Variant Calling of SNVs and small InDels (in .vcf format, see Table 2 and Table 3 )
Identify single nucleotide variants (SNVs) and small insertions/deletions (InDels < 50 bp) with variant calling results available in .vcf format. - Annotation of Variations with Amino Acid-Level Consequences (in .html format, see Table 4)
Understand variations with potential amino acid-level consequences, presented in .html format (accessible for most model organisms/if the reference genome is annotated accordingly).
Optional:
- Filtering of Variants Against a Background Sample (in .html format)
Customize your analysis by filtering variants against a background sample, with results provided in .html format. - Sample Consensus Sequence (in .fasta and .gb format)
Receive a sample consensus sequence, potentially resulting in a chimeric sequence, in .fasta and .gb formats.
Complementary Bioinformatic Analysis (at additional cost)
Genomic Epidemiology (for prokaryotes only):
- Screening for matches to resistance and virulence databases and mycotoxin genes (in .tsv format)
Identify matches to resistance and virulence databases with screening results provided in .tsv format. - Phylogenetic Typing of Sample Strain (in .pdf format)
Understand the phylogenetic relationship of your sample strain through a concise .pdf report.
These results empower you with a comprehensive view of your re-sequencing analysis, enabling you to extract meaningful insights and make informed decisions.

Table 1: This detail of a result table shows detected single nucleotide variations, small insertions and deletions and their annotation.


Table 3: This detail of a result table lists putative structural variations.
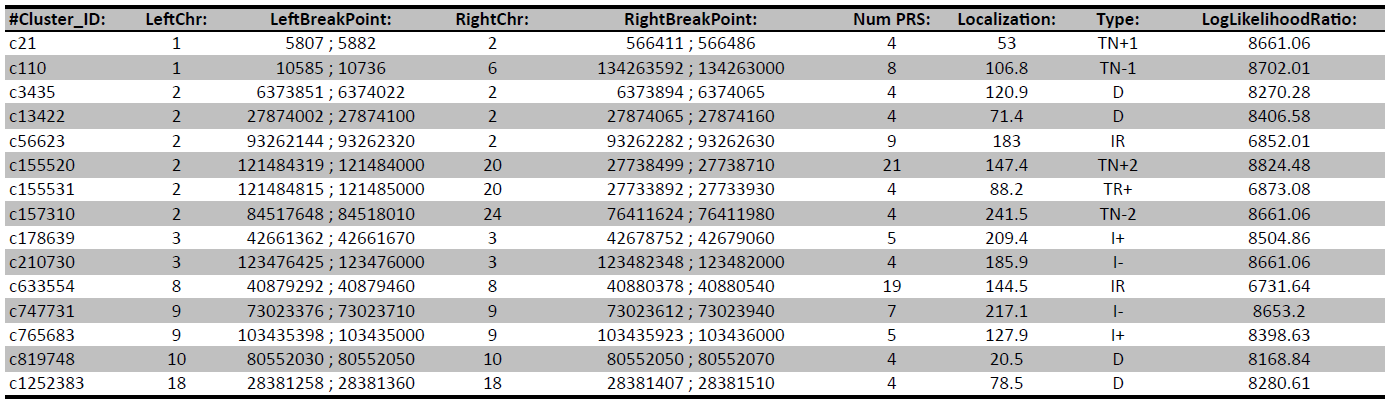
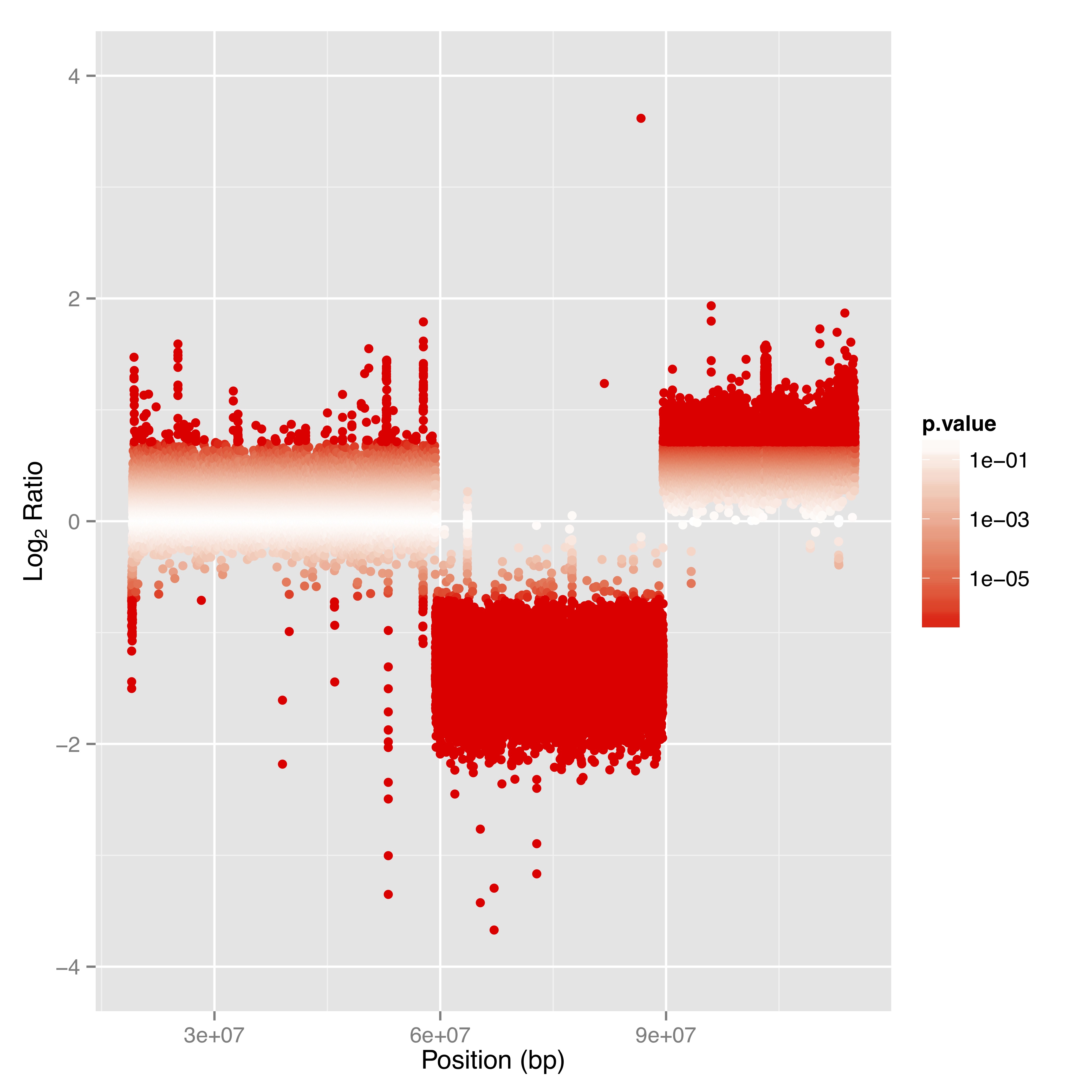
Figure 2: This figure depicts a possible copy number variation in relation to a reference sample.
Table 4: Table displaying detailed information on each observed variation like variant type and position, affected protein including reference and altered sequence.

Turnaround Time
- Delivery of data within 20 working days upon sample receipt (includes library preparation and sequencing)
- Additional 10 working days for data analysis (bioinformatics)
- Express service possible on request